Gene Selection
The advancement of
bioinformatics has absorbed tremendous attentions in the last decade. Using
modern microarray technology, great achievement has
been made in medical and biological researches. Although the progress in microarray technology is prominent, there are still many unresolved
problems hindering scientists making more progress by microarray
data. The unbalance between the large number of genes tested on microarray chip and the relative small number of samples
can be one of the most challenging problems. In microarray
experiments, we are already able to monitor the expression level of hundreds
and thousands of genes simultaneously. However, the sample size is significantly
smaller than the gene size. To process such a large number of data is
time-consuming and inefficient. Thus, it is necessary to select the
"important" genes from the gene dataset to reduce the dimension of
the dataset, to generate classifier and prepare for the clustering procedure.
There are tons of
genes in mammalians that controls the protein metabolism and modulates the
function of the body. Some genes keep their expression level constant under
different conditions. These genes are called house keeping genes. They are not
important genes we are looking for to construct classifier. Other genes, on the
contrary, have different expression levels under different conditions and in
different tissues. These differentially expressed genes are called
discriminatory genes. They contain valuable information that is related to the
features of the samples. This step of discriminatory gene identification is generally
referred to as gene selection. Gene selection is a pre-requisite in many
applications [8]. It should be noted that, often, the number of unrelated genes
is much larger than the number of discriminatory genes.
Gene selection is
to detect a subset of a small number of discriminatory genes that can
effectively recognize the class to which a sample belongs. That is, the
classification error is minimized by given classifier. The known dataset that
is used for learning the classifier, or equivalently for gene selection, is
referred to as training dataset. In a training dataset, every sample is labeled
with its known class. Notice that sometimes a class is significantly larger
than the others, then the trained classifier might be
biased. Therefore, the desired gene selection methods are those that are not
affected by the sizes of classes in the training dataset. After picking the a subset of a small number of discriminatory genes,
these number of genes will be used on the testing samples. Note that testing sample
is different from the training samples. The classification accuracy is defined
as the percentage of correct decision make by the classifier on the testing
samples. Good gene selection method can obtain the better classification
accuracy.
Another major
concern for gene selection and classification is over-fitting. Over-fitting
refers to the creating diagnostic models that may not generalize well to new
data from the same sample types and data distribution despite good performance
on the training set [12]. It is very easy to over-fit the classifiers and the
gene selection procedures, because a lot of algorithms are highly parametric
and datasets consist of a relatively small number of high-dimensional samples,
especially when using intensive model search and powerful learners [12].
Another problem is under-fitting, which leads to an inefficient classifier that
is not optimally robust due to limited experimentation [12]. Under-fitting is
easy to happen under some circumstances, such as the application of a specific
learning algorithm without consideration of alternatives, or the use of
parametric learners with certain values of parameters without searching for the
best ones [12].
In this survey, we
intend to survey recent progress in Gene selection research, to have a better
picture of whole work, Section [2] investigate some the latest techniques
developed for gene selection.
There are a
variety of gene selection methods proposed in the last a few years, some of
which are based on some statistical models. Xiong et
al. [4] suggested a model to select genes through the space of feature subsets
using the classification error. Guyon et al. [6] proposed
a gene selection approach utilizing support vector machines based on recursive
feature elimination. These gene selection methods do not assume any specific
data model for the gene expression data. However, it is reported [13] that
their results may be influenced by the specific classification methods used for
scoring the genes.
Another category
of widely used gene selection methods is through model-based probabilistic
analysis. For example, Baldi et al. [1] developed a
Gaussian gene-independent model to process the gene expression data. They
implemented a t-test combined with a full Bayesian treatment on the gene
expression data. A disadvantage of this category of gene selection methods is
the lack of adaptability, because it is unlikely possible to construct a
universal probabilistic analysis model that is suitable for all kinds of gene
expression data, where noise and variance vary dramatically across different
gene expression data [13]. Thus, model-free methods are more desirable.
In the following
part, we will review some details on several classical and latest gene
selection methods and their corresponding score function.
Assume there are a total p genes in the microarray chips, and assume we have in total n samples
that are grouped into L classes in the training dataset. Let aij denote the expression value of
gene j in sample i. The training
dataset can thus be represented as a matrix
![]()
Let nk denote the number of samples in class Ck,
for k = 1, 2, ..., L (i.e., ![]() ). Let
). Let ![]() , which is the average expression value of gene j in
class Ck, for k = 1, 2, ..., L. The expression vector
, which is the average expression value of gene j in
class Ck, for k = 1, 2, ..., L. The expression vector ![]() is the centroid of class Ck. Correspondingly,
the centroid matrix is
is the centroid of class Ck. Correspondingly,
the centroid matrix is
![]()
The mean of centroids is ![]() , where
, where ![]()
For sample i belonging to class Ck, the
difference between the expression value of gene j and the average
expression value in class Ck is ![]() . The matrix
. The matrix
![]()
is the variation matrix of the
training dataset. Let ![]() denote the average
variation for samples in class Ck with respect to the centroid. The centroid variation
matrix is
denote the average
variation for samples in class Ck with respect to the centroid. The centroid variation
matrix is
![]()
Intuitively, most
of methods assume the feature genes have small intra-class variation
and large inter-class variation.
F-test method [3]
is also a single gene scoring approach. Besides the notations used in our
methods, it is a classical gene selection method. It uses ![]() to denote the
variance of expression value of gene j in the k-th class:
to denote the
variance of expression value of gene j in the k-th class:
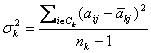
and ![]() to denote the
variance in the whole dataset. Gene j has a score defined to be:
to denote the
variance in the whole dataset. Gene j has a score defined to be:
![]()
3.2 Cho’s Method
Using the same
notations as above, Cho's method [2] defines a weight
factor wi
for sample i, which is ![]() if sample i belongs to class k. Let
if sample i belongs to class k. Let ![]() . The weighted mean(j)
for gene j is defined as
. The weighted mean(j)
for gene j is defined as
![]()
The weighted
standard deviation is defined as
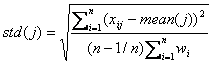
Then the score of
gene j is calculated as
![]()
where ![]() is the standard
deviation of centroid expression values
is the standard
deviation of centroid expression values ![]() .
.
Both GS1 and GS2 [15]
define the scatter of gene j to capture the inter-class variations,
which takes in ![]() as a component:
as a component:
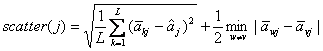
in which the square root is the standard
(estimated) deviation of all the centroids on gene j.
For the intra
class variation, GS1 choose the funtion compact1(j)
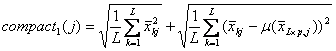
and GS2 choose the function compact2(j)

as the intro-class score for gene j. Therefore,
![]() are the score function for GS1 and
GS2 respectively.
are the score function for GS1 and
GS2 respectively.
SFS(Sequential forward search) and
SFFS(sequential forward floating search) algorithms were proposed by Pudil et.al.[10], and was
employed in Xiong et.al's
feature selection article [14]. SFS first find the most significant feature and
add it into selected feature pool. Then SFS find the feature which leads to the
most significant combination with the feature/features in the selected feature
pool. One problem of SFS is that once a feature is added into selected feature,
there is no chance that it would be removed out. However, SFFS tries to solve
this problem. It not only tries to find the best feature combination, but also
tries to remove features in the selected feature pool to see if that increases
the significance.
There are a
variety of algorithms proposed to devise classifier. The most widely used
classifier is support vector (SVM) machine-based classification method. SVMs have achieved excellent classification performances in
most tasks [12]. The advantage of these approaches is that they are insensitive
to the problem of dimensionality and are able to deal with large-scale
classification in both sample and variables [12]. The early stage SVM can only
handle classification for two classes, which is called Binary SVMS. The basic
idea of binary SVMS is to use a kernel function to implicitly map data to a
higher dimensional space, and to identify the maximum-margin hyperplane that divides training instances [12]. In 1999, Kressel proposed the simplest multiclass
SVM method, called one-versus-rest (OVR) method, which constructs k binary SVM
classifiers [12]. He also proposed another multiclass SVMs at the mean time,
which is one-versus-one (OVO) SVM classifier [12]]. These two methods made
tremendous progress in the development of SVMs, but they
also have apparent limitations. These approaches are computationally
non-economic, and do not incorporate analysis of generalization which is
necessary for a good learning algorithm [12]. Later on, other researchers have
made modifications with SVMs and improved the classifier.
Platt et al devised classifier named DAGSVM (2000) [9]. Hsu and Lin (2002),
Weston and Watkins (1999) also proposed their classification methods based on
the SVMs [7]. In addition to SVMs,
K-nearest beighbors (KNNs)
is another robust classifier. A KNN classifier defines the class of a testing
sample by analyzing its K nearest neighbors in the training dataset [3].
In this method,
samples are regarded as points in m-dimensional space, and then Euclidean
distance or other distance calculation is used to classify the samples by a
vote of K-nearest training instances [12]. Another classification method is backpropagation neural networks (NNs).
The network is composed of three parts: the input layer of units, the hidden
layer(s) of units, and the output layer of units [12]. The algorithm adjusts
the weights between units in the network until a vector of weights is found
that best fits the training data [12]. After the training session is finished,
the testing data can be imported into the input layer, and the network will
output the class the sample belongs to by output layer [12].
Probabilistic
neural network (PNNs) is a member of Radial Basis
Function (RBF) neural networks methods [16]. PNNs
have a very similar structure with NNs, comprising
three layers. However, the PNNs can only have one
hidden layer, while PNNs can have more than one. The
difference between these two methods is that for PNNs,
the inputs pass directly to the hidden layer with no weights [12]. The PNNs use Gaussian density function as an activation
function for the hidden layer and a least squares optimization algorithm is
used to calculate the weights for the connection between hidden layer and
output layer [12].
Among the various
classification methods, SVMs and KNNs
are most widely used. When using classifiers, a few problems have to be noticed
in order to obtain the best classification performance. First is the
combination of algorithms. For different dataset, we can combine specific gene
selection methods and classification methods. Different selection or
classification methods can also be grouped together to improve the selection
efficiency and classification accuracy. Statnikov et
al found that combining classifier Decision Trees (DT) and MC-SVM can yield
good classification accuracy [12]. We also found that combining the clustering
of genes and other general gene selection method can improve the quality of the
genes selected, and achieve better classification results.
The classification
of cancer has been one of the most successful examples for the application of
classification. The use of classification in cancer research is prominent
because of the variety of cancer types. The different types of cancer require
different treatment. However, the traditional diagnosis by histopathological
analysis has a high chance of mistake.
The gene
classification can be used not only for class prediction, but also for class discovery
prediction [3]. Class discovery refers to identifying new tumor subtypes, and
class prediction is to assign the particular tumor samples to existing classes,
which could reflect current states or future outcomes [5]. Using microarray data, Golub et al
successfully discovered the distinction between acute myeloid leukemia (AML)
and acute lymphoblastic leukemia (ALL) without
previous knowledge of these cancer types [5]. Fu et al applied a gene
classification method on small round blue cell tumors and leukemia microarray dataset and compare the classification accuracy
with results by artificial neural networks and statistical techniques [4]. They
demonstrated the reliability of their method by the consistency with different methods
and other biological evidences [4]. These studies confirm our confidence in the
prospect of using molecular technique of microarray
in cancer diagnosis.
Recently we found
another way to improve the accuracy of gene selection is to select genes from
different groups. Because most of the current algorithms only use a limited
number of discriminatory genes to device the classifier, the quality of the
genes selected is essentially important. We select genes from different groups,
which maximize the dimension of the discriminatory gene set, and enhance the
accuracy of the classification. After gene selection, a classifier will be
devised based on discriminatory genes. The classifier then will cluster the
samples by its microarray dataset. (Delete this
sentence)
To explain the way
we partition genes, we need to introduce the basic idea of genetic network
here. A single gene could modulate (activate or inhibit) some genes, and at the
same time, be modulated by other genes. The modulation of one gene to another
could be condition dependent and tissue specific. Some genes who
share the same expression pattern, the same function, or co-express in specific
tissue or under some conditions, can be grouped together as a module. Then we
find the expression pattern of these modules in microarray
dataset, that is to match the condition or tissue with
the module that has significantly higher or lower expression pattern. By doing
so, the gene modules can be connected with specific condition or tissue type.
Finally, biological evidences of the regulatory relationship can be added to
different genes and modules to reflect the complex regulatory relations in the
network. In a typical genetic network, gene is represented by vertices, and
edge between genes stands for a regulatory program. The value of the edge
(either 1 or 0) stands for activating or inhibiting. The network can also be
used to discover few relationships between conditions and genes, or between
genes themselves. For example, Segal uses a network with 2849 genes and found
that activation of some modules is specific to certain types of tumor, for
example, a growth-inhibitory module is specifically repressed in acute lymphoblastic leukemias and may
contributes to the deregulated proliferation in these cancers [11].
For the grouping,
we can assume the genes in different modules represent the same pattern of
expression, signaling pathway, or same function. Genes in the same module may
play the same role in classification. Thus we limit the number of genes being
selected from the group or module to enhance the dimension of the genes and
improve the accuracy of classification.
[1]
P. Baldi and A. D. Long. A Bayesian framework for the analysis of microarray expression data: Regularized t-test and
statistical inferences of gene changes. Bioinformatics, 17:509{519, 2001.
[2]
J. Cho, D. Lee, J. H. Park, and
[3]
S. Dudoit, J. Fridlyand,
and T. P. Speed. Comparison of discrimination methods for the classification of
tumors using gene expression data. Journal of the American Statistical Association, 97:77{87, 2002.
[4]
L. M. Fu and C. S. Fu-Liu. Multi-class
cancer subtype classi¯cation based on gene expression
signatures with reliability analysis. FEBS Letters, 561:186{190, 2004.
[5] T. R. Golub. Molecular classification of cancer: Class discovery
and class prediction gene expression monitoring. Science, 286:531{537, 1999.
[6]
[7]
C. W. Hsu and C. J. Lin. A
comparison of methods for multi-class support vector
machines. IEEE Transactions in Neural Networks, 13:415{435, 2002.
[8]
S. Mukherjee and S. J. Roberts. A theoretical analysis of gene
selection. In The Pro-ceedings of IEEE
Computer Society Bioinformatics Conference (CSB 2004), 2004.
[9]
J. Platt, N. Cristianini, and J. Shawe-Taylor. Large margin dags
for multiclass classification. Neural Information Processing
Systems, 12:547{553, 2000.
[10] P. Pudil, Novovicova. J., and J Kittler. Floating search methods
in feature selection. Patt. Recogn. Lett, 15:1119{1125, 1994.
[11]
E. Segal, N. Friedman, D. Koller, and A. Regev. A module
map showing conditional activity of expression modules in cancer. Nature, 36:1090{1098, 2004.
[12]
A. Statnikov, C. F. Aliferis,
[13]
O. G. Troyanskaya, M. E. Garber, P. O. Brown, D.
Botstein, and R. B. Altman.
Non-parametric methods for identifying differentially expressed genes in microarray data. Bioinformatics, 18:1454{1461, 2002.
[14]
M. Xiong, X. Fang, and J. Zhao. Biomarker identi¯cation
by feature wrappers. Genome Research, 11:1878{1887, 2001.
[15] K. Yang, Z. Cai, J. Li, and G. Lin. A model-free and stable gene selection in microarray
data analysis. In The
Proceedings of IEEE The 5th Symposium on
Bioinformatics and Bioengineering (BIBE 2005), pages 3{10, 2005.