3 Missing
Value Estimation Methods
3.1 Weighted
K-nearest Neighbors (KNN) Method
3.2 Sequential
K-nearest Neighbors (SKNN) Method.
3.3 Bayesian
Principal Component Analysis (BPCA) Based Method
3.4 Local
Least Square Imputation (LLSimpute) Method
3.5 Iterated
Local Least Square Imputation (ILLSimpute)
4 Measures
of Performance of Imputation Methods
3.1 Root
Mean Square Error (RMSE)
3.2 Normalized
Root Mean Square Error (NRMSE)
3.3 Gene
Selection Based Measuring Method
As one of the most
important research tool in bioinformatics, microarray
has made substantial advancement in recent years. Combining with the
mathematical, electronical and biological techniques,
Microarray provides a useful tool to monitor the expression
level of large amounts of genes simultaneously. Using microarray
technology, biological researchers has harvested tremendous data in gene
expression studies and hastened the paces of biological studies. Meanwhile,
researchers in mathematics and computing science have developed various
algorithms, and made contribution to both microarray
data processing and theoretical mathematics studies. Following is a brief
introduction of the methodology of microarray.
Firstly, cells from different tissue or different source are treated with
certain condition before biological digestion. After that, mRNA is extracted
and hybridized with microarray chip. The intensity of
the fluorescence on the chip will represent the expression level of the gene on
the spot. Microarray experiments usually generate
data in the format of a matrix, with rows representing different genes, and
columns representing different samples and conditions. After the matrix dataset
is obtained, mathematical transformation and algorithms can be applied to the
dataset, to extract useful information and underlying pattern. Current popular microarray data analysis methods
include missing value estimation, feature selection, clustering and gene
network construction. This survey focus on missing
value estimation, the primary step, and reviews its development in recent years
and current proceedings.
Missing value can
originate from different any stage of experiment, from chip production and
treatment, hybridization and scanning [5]. A number of reasons could cause
missing data, to name some, insufficient resolution, image corruption, or even
dust and scratches on the slide [1]. Chips with probes packed too tightly are
more possible to produce missing values. In these cases, values are missing at
random. Signals whose intensities are too low to recognize or measure are
eliminated as missing values too. These values are considered as non-random
missing value [5]. It is introduced above that a number of algorithms have been
developed to explore the underlying pattern of microarray
data, such as gene selection and classification. However, these algorithms must
apply on complete datasets. Therefore it is crucial to obtain accurate and
complete dataset. One possible solution to recover the dataset with missing
value is to repeat experiments, which is inefficient and infeasible. Hence, the
missing values in original data need to be predicted mathematically and
estimated data must be put in before any other algorithms are applied.
3
Missing Value Estimation Methods
Predicting missing
values with accuracy assures the reliability of the results from subsequent
steps. At beginning, simple imputations were used to, e.g., filling the missing
values with zeros; using the row mean for imputation. These methods produce inaccurate
estimating values and influence the results of gene selection and classification.
Then a number of complicated approaches have been proposed to predict missing
values. This survey will review popular algorithms at present. We will introduce
the classic methods and methods derived from classic ones.
3.1 Weighted K-nearest
Neighbors (KNN) Method
Troyanskaya et al. proposed that the KNN-based
method takes k nearest neighbor genes from the dataset except genes that
have missing value at the same position with the target gene (gene whose
missing value is being estimated). Euclidean distance is used to calculate the
similarity between target gene and neighbor genes [6]. After the neighbor genes
are selected, a weighted average of values from k nearest genes is then used as
an estimate for the missing value in target gene. The calculation of weight of
each gene can as following equation.

where k is the number of selected genes and Di is the distance between ith
gene and target gene.
In this method, missing
values in the same gene need to be calculated separately. Troyanskaya
et al. examined this method and showed that this method was efficient at
predicting missing values. It is also suggested that KNN method works better on
non-time series data and time series data [6].
3.2 Singular Value
Decomposition Based Method (SVDimpute)
SVDimpute was proposed by Troyanskaya
et al. too [6]. Overall, SVDimpute performs
worse than KNN method. However, it works better on time-series data than KNN.
In SVDimpute method, a set of mutually orthogonal
expression patterns are obtained and linearly combined to approximate the
expression of all genes, through the singular value decomposition of the
expression matrix Gm*n. By selecting k most significant eigengenes, a missing value gij
is estimated by first regressing the i-th gene against these k eigengenes
and then using the coefficients of the regression to reconstruct gij from the linear combination of the k eigengenes
3.3 Sequential K-nearest
Neighbors (SKNN) Method
Kim et al.
improved KNN method and proposed SKNN method in 2004 [3]. The difference
between SKNN and KNN is
1.
The original data set is divided into complete dataset and incomplete
dataset, according to if the gene has missing value or not. Genes in incomplete
dataset is ordered according to its missing rate (how many missing values it
contains).
2.
Imputation starts from the gene with least missing rates. k nearest neighbor genes are selected from complete
dataset. Once a gene has all missing values estimated, it will be moved to
complete dataset for the imputation of next gene. In this method, missing
values in the same gene can be calculated simultaneously.
Information
carried by genes with missing valued is better used than KNN method. Kim et
al. also demonstrated that SKNN method performs better than KNN especially with
data carrying large amount of missing values [3].
3.4 Bayesian Principal
Component Analysis (BPCA) Based Method
Bayesian model is
a popular model which has been widely used. However, Oba
et al. first applied this model in missing values estimation [4]. BPCA
method mainly comprise three elementary processes, which are
1.
Principal component regression.
2.
Bayesian estimation.
3.
Expectation-maximization-like repetitive algorithm.
The performance of
BPCA method has been compared with that of KNN and SVDimpute
and showed its advantages over them [4]. However, when genes have dominant
local similarity structures, BPCA doesn't work as well as KNN [4].
3.5 Local
LLSimpute method is a novel method published in 2005
[3]. In this method, the first step is to select k genes by Pearson correlation
coefficients or Euclidean distance [3]. Second, missing values in the target
genes are estimated by local least squares. The key step in LLSimpute
is using local least squares to determine the coefficients to approximate the
target gene. The target gene is assumed to have a linear combination with its
coherent genes.
Without
loss of generality, we assume that the target gene is gene 1 and it has missing
values at the first n-nˇŻ positions. Suppose there are k coherent genes for gene 1
and they are genes s1, s2,
ˇ, sk. We select the rows 1,
s1, s2, ˇ, sk from matrix Gm*n to
compose the following sub-matrix:
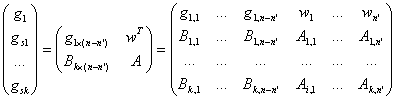
The k-dimensional
coefficient vector x* is the one that minimizes square![]() , that is,
, that is,
![]() .
.
The target gene is
approximated as
![]() ,
,
and the missing values in the target gene as
estimated as
![]() .
.
H. Kim et al.
[3] showed that the LLsimpute performs better than
all the previous methods.
3.6 Iterated
Cai et al. improved LLSimpute
in two steps. First, instead of set k as a fixed number of neighbor genes, the
average distance is calculated and we learn a distance ratio, and the genes
with their distances not exceeding times the average distance are considered as
neighbor genes to the target gene. The distance ratio is the ratio that
achieves the best accuracy in training dataset. In this method, the number k
will vary for different target genes. Second, after the LLSimpute
method is run to estimate the missing values, the algorithm use the imputed
results from the last iteration to re-select coherent genes for every target
gene, using the same distance ratio, and runs LLSimpute
method to re-estimate the missing values. The process iterates a number of
times or until the imputed values converge [1].
As we have
introduced above, each method has its advantage and disadvantage, and can be
associated with a specific type of systematic error [2]. Therefore, Jornsten [2] and colleagues proposed a new method called LinCom, which is the convex combination of the estimates by
ROW, KNN, SVD, BPCA and GMC [2]. LinCom is especially
useful to understand the merits of different imputation [2]. Jornstern et al. inferred that by combining the
various estimates, they could borrow strength across methods to adapt to the
data structure. As single missing value estimation method is still improving,
and new methods coming out quickly, combination of methods can be another
avenue to generate more efficient and accurate tool for missing value prediction.
4
Measures of Performance of Imputation Methods
There are two
standard measurements in imputation methods which are root mean squared error
(RMSE) and normalized root mean squared error (NRMSE).
4.1 Root Mean Square Error
(RMSE)
RMSE is commonly
methods to measure the performance of missing values. Generally, some
pseudo-missing values are generated to test the missing value estimating methods.
The estimated values are compared with true values. Let X = {x1,
x2, ˇ, xq}
be the set of missing value positions and assume the true expression value at xi
is ai while the estimated value is ai*. Define
![]()
to be the mean of all squares of errors. Define
![]()
to be the mean and deviation of all the true
expression values, respectively. RMSE is calculated as following [2]:
![]()
4.2 Normalized Root Mean
Square Error (NRMSE)
When all missing
values have been estimated, the quality of the imputation also can be measured
by normalized root mean squared error (NRMSE) [3]. Then, the NRMSE is
defined as
![]()
Essentially, the
NRMSE indicates how close the estimation values and the true values are. Therefore,
the smaller the NRMSE value, the better quality the imputation has.
4.3 Gene Selection Based
Measuring Method
Scheel et al. indicated that since missing
value estimation served the purpose of further analysis such as gene selection,
it would be helpful to evaluate the performance of missing value estimation methods
by their effect on gene selection results [5]. They measured the score of
missing value estimation by looking to lost and added differentially expressed
genes compared with the list of differentially expressed genes from an analysis
of the true complete data [5].
As one of the most
promising technique in recent years, microarray has
been attracted more and more attentions. The large amount data it produces
bring both chance and challenge. The processing and analysis of microarray becomes a fascinating topic for scientists from
computing science, statistics and mathematics. Missing value estimation is the
first and essential step in microarray data processing.
A number of good models and algorithms have been proposed to accurately predict
missing values. We reviewed the development of missing value estimation
methods, and summarized the current popular method. We also compared the
performance of these methods, introduced their principles, advantages and disadvantages.
At last, we briefly introduce the commonly used methods to measure the
performance of missing value estimation.
[1] Z. Cai, M. Heydari,
and G. Lin. Microarray missing value imputation by
iterated local least squares. In The
Proceedings of Proceedings of The Fourth
Asia-Pacific Bioinformatics Conference (APBC 2006), 2006.
[2]
R. Jornsten, H. Wang, W. J. Welsh, and M. Ouyang. Dna microarray
data imputation and significance analysis of differential expression. Bioinformatics, 21:4155¨C4161, 2005.
[3] H. Kim, G. H. Golub, and H. Park. Missing value extimation
for DNA microarray gene expression data: Local least
squares imputation. Bioinformatics, 20:1¨C12, 2005.
[4] S. Oba, M. Sato,
[5]
[6] O. Troyanskaya, M. Cantor, G. Sherlock, P. Brown, T. Hastie, R. Tibshirani, D.
Botstein, and R. B. Altman. Missing value estimation methods for dna microarrays.
Bioinformatics, 17:520¨C525, 2001.