Microarray Normalization
3 Current Normalization
Methods
3.1 Within Slide Normalizations
3.2 Multiple Slides Normalizations
4 Evaluation Models for Normalization Methods
4.1 Ration-Intensity plot (R-I
plot)
4.4 Comparison Using Co-expression Genes
4.5 Comparison of Different Normalization Models
Every DNA microarray experiment will generate a comprehensive dataset
in the format of matrix to express the DNA levels of a population of cells
under a certain experimental condition [5]. In the matrix dataset, normally
the expression levels of genes are specified in rows, and the experimental
conditions ordered in columns [3]. In order to abstract useful and reliable
information from the dataset, mathematical methods are involved in the data
analysis process. Before any algorithms can be applied to survey the underlying
gene expression pattern from original data, a very important pre-process
procedure called normalization must be performed to raw data. In the following
sessions, we will discuss the necessity of normalization,
review the current methods for microarray data
normalization, and for normalization results evaluation. Finally, we will
compare and comment the available normalization methods.
Microarray is usually used to detect
the expression level variation between different samples. Gene expression
level comes from the intensity of fluorescence detected on the chip after
hybridization. The intensity can be affected by various reasons, to name
some, the amount of RNA in the sample, the hybridization quality, and detection
system. A lot of reasons can lead to inconsistency between samples, which
make normalization necessary, e.g., unequal quantities of starting RNA,
differences in labeling or detection efficiencies between the fluorescent
dyes used, and systematic biases in the measured expression level [6]. Therefore,
it is important to standardize the original values for meaningful comparison
between samples to be made. Normalization is the transformation of data
in order to eliminate questionable or low-quality measurements, to adjust
the measured intensities to facilitate comparison, and to select genes that
are significantly differentially expressed between classes of samples [6].
3 Current Normalization Methods
There exit various normalization
methods, which are based on different rationales of solving different problems
in microarray data analysis. The relations between
these normalization methods are not quite straightforward and there are
still new normalization methods coming up, but basically these methods can
be classified as illustrated in Figure 1.
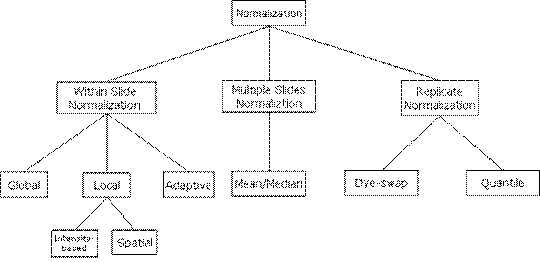
Figure 1: Normalization
Methods Classification.
Normalization methods
can be mainly classified into three classes, Within Slide Normalization,
Multiple Slides Normalization and Replicate Normalization. Within Slide
Normalization methods try to eliminate or reduce the variances among gene
spots within a single microarray slide while Multiple
Slides Normalization methods reduce the variance between multiple microarray slides. Replicate Normalization methods apply to
replicate experiments. The following article of this section introduces
detailed methods under each main normalization class.
Before we get into each method, let us get familiar with some basic knowledge
of microarray data transformation. As mentioned
above, most microarray experiments are for comparison
purpose. For each single gene, we record its intensity ratio of sample R
and G (R and G represents Red and Green fluorescence
respectively) as T. Therefore we have following definition: Ti = Ri/Gi.
Ri and Gi are measured intensity
of ith gene. The ratio provides us a good
measure of expression variation, however, the problem
with it is that it treats up-regulated and down-regulated genes differently
[6]. Then the ratio is transformed by logarithm base 2, which produces a
continuous spectrum of values and treating up-regulated and down-regulated
genes in a similar fashion [6]. This transformation method is prevalently
used in most normalization techniques.
3.1 Within Slide Normalizations
Because there are always variances among different gene spots within one
slide and the intensity of different fluorescent dyes are always not the
same which contradicts common assumption, Within Slide Normalization methods
are proposed to eliminate or reduce these kinds of variances. Under the
Within Slide Normalization branch, there are basically three methods, Global,
Local (Intensity-based, Spatial) and Adaptive.
For this normalization method, people always assume that equal amount of
sample RNA is used in microarray experiment to
be compared. Given the fact that there are millions of RNA molecules in
the sample, and the average mass of all the molecules is about the same.
Thus, if the genes on the chip are randomly selected, we will expect same
or similar amount of genes hybridized to the chip, that is, total intensities
of all the genes on the chip are the same. Notice that, this method does
not apply to the situation where the genes on the chip are not randomly
selected, and it is expected one sample will produce more intensity than
another. We will talk about this in detail in Adaptive Normalization method.
Using the above rationale, we sum up the intensities of the hybridization
with each sample, and calculate the ratio between them [6]:
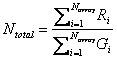
For each gene or element in the array, we have
![]() . After transformation of logarithm, we have log2(T0 i
)
= log2(Ti) ¡ log2(Ntotal).
. After transformation of logarithm, we have log2(T0 i
)
= log2(Ti) ¡ log2(Ntotal).
Aside from global variance between different fluorescent dyes, there also
exist intensity variances among different gene spots. They are mostly intensity-based
variance and spatial variances. Therefore, intensity-based normalization
and spatial normalization are introduced to get rid of these two variances.
Intensity-based and Spatial Normalization
It has been reported that the log2(Ti) is systematically intensity-dependent, which is most common for low density spots [6]. The normalizations which normalize these kind of biases are called intensity-based normalizations. Normally, intensity-based normalizations correct the lowess deviation through a weighted linear regression as a function of the log10(intensity) and subtracting the calculated best-fit average log2(ratio) from the experimentally observed ratio for each gene [6].
Within microarray slide, the locations of gene spots will also affect the
gene expression
![]()
where
M = log2(R/G) and A = (log2R + log2G)/2
N is the final corrected intensity value. loess(r, c) is a two-dimensional lowess function of the row position r and the column
position c of the spot on the array. loess(A)
is the global lowess function which gives the
correction value of M based on intensity A. There are some
other lowess methods, but the above one is representative because
it not only include intensity-based normalization
but also include spatial normalization.
Intensity-Based Filtering of Array Elements
In a microarray experiment, the detection of low
intensity is more likely to be inaccurate. For high intensity spots, those
that exceed the saturation lack accuracy, too. Therefore, sometimes it is
necessary to perform normalization methods to filter the extreme data. A
few approaches have been used to deal with such problems. For example, those
intensities exceeding saturation could be eliminated, setting a threshold
as the high limit. Similarly, a low threshold can be set as well. Another
approach is called percentage-based cut-offs, in which a certain percent
of lowest intensities and/or highest intensities are removed [6]. Statistics
can also be used to filter the extreme data. A confidence level can be set
to eliminate too deviant data.
Most normalization methods like global normalization, intensity-based normalization
and spatial normalization assume that most genes on the microarray slide are not differentially expressed between
the two hybridized samples and that for the differentially expressed genes,
the direction of the difference is symmetric between the two samples [8].
However, considering the following three cases, these assumptions are not
appropriate: 1: more than half of the genes are differentially expressed
on the array; 2: the numbers of over- and under-expressed genes on
the array are unequal; 3: only genes of specific biological interest
are selected to make a customized array, which are highly variable across
the samples. Therefore, Yingdong
Zhao et al. generalized Newton et al.'s Gamma-Gamma-Bernoulli
model and proposed an adaptive method based on three-component mixture model
for normalization of dual labeled microarray data. Their results show that the performance of
this adaptive normalization method overwhelms global method and lowess method.
3.2 Multiple Slides Normalizations
If comparison of gene expression values from multiple microarray
slides is needed, these expression values need to be scaled to the same
level for further analysis. Therefore, normalization over these different
slides is required. We call this normalization scale-normalization. Scale-normalization
is a simple scaling of the M-value from a series of arrays so that each
array has the same median absolute deviation [7].
Suppose we use median scale-normalization. The scaled M (log2(T0 i
))
value is
![]()
Ti = Ri/Gi
meank means the mean value
of the kth slide and median means
the median value of all the slides' mean M value.
Although it is always expensive, replicate experiments can generate a set
of expression values for each gene spot. Using the mean or median of each
gene's data set is a common method. However, to utilize the replicate experiment
data, two different normalization methods are proposed, Dye-swap Normalization
and Quantile Normalization.
Because different fluorescent
dyes may have different effects in a single slide. To minimize the bias
caused by the fluorescence labeling, the method of swapping fluorescent
dye and replicating the experiment on the same sample is proposed. After
the replicating is performed, values from replicated experiments need to
be averaged. Following equation will produce the average value for replicates:
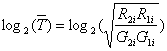
3.3.2 Quantile Normalization
This normalization method
is propose by Bolstad, et al. [2] The rationale of this method is to make the distribution of
the intensities for each array the same in a set of arrays. Following is
the procedures proposed by [2]
l
Given
n array of length p, form X of dimension p*n where each array is a column
Sort each column of X to give Xsort
l
Take
the means across rows of Xsort and
assign this mean to each element in the row to get X’sort
l
Get
Xnormalizaed by rearranging each column of X’sort to have the same ordering as
original.
The following example illustrates
a procedure of how Quantile Normalization works.
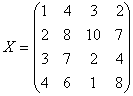 ,
,
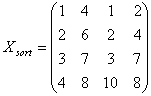 ,
,
 ,
,
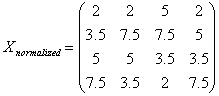
The problem with this method
is it assumes the values across all the arrays are same, which is not true.
However, because a single gene usually has replicates in an array and the
averaging will compensate for the inaccuracy produced by the assumption.
It is worth mentioning here, most of the normalization methods can be applied
either on the whole dataset or a specific sub-region in the array to adjust
regional bias.
4 Evaluation Models for Normalization Methods
4.1 Ration-Intensity plot (R-I plot)
R-I plot (Ration-Intensity
plot) is a diagram to show the ratio and intensity relation of each element
in an array. The horizontal axis is log10(Ri*Gi), and the vertical axis is log2(Ri-Gi). The
range of vertical axis usually centers on log2(Ri-Gi) =
0. Good normalization method will adjust the data so that the ratio will
center around log2(Ri-Gi) = 0.
Variance comparison is
used to compare the results of two normalization methods on certain datasets.
For a subset of probes in the array, the expression values are normalized
by different approaches, and then the mean value and variance are calculated.
The mean and variance of a subset are plotted in the diagram. The horizontal
axis is the logarithm of mean, and the vertical is the logarithm of the
variance ratio (log variance1/variance2). By this plot, we can compare the
variance of the data after different normalization method. In the diagram,
the plot with its loess smoother above the log variance
= 0 means the first method produce larger variance than
the second one. The plot with its loess smoother below the log (variance)=0 means the first
method produce smaller variance than the second one. The normalization method
that produces least variance is better.
Bias comparison is an approach
to detect bias using dilution-series samples. Using samples with different
concentration, we expect the expression value E and concentration c can
fit into the following model:
![]()
Since the intensity depends
on the amount of corresponding RNA, ideally β1 is close to 1. Using the data
applied with different normalization methods, models can be estimated and
value can be compared. The normalization method that produces the model
with the β1 value closest
to 1 performs best to adjust the expression value and concentration relationship.
There are a variety of other methods to evaluate the normalization methods.
For example, Bolstad et al. [2] proposed
a method to compare the ability the normalization methods to reduce the
pairwise differences between arrays. He used the
average absolute distance from loess smoother to log2(ratio) in
R-I plot.
4.4 Comparison Using Co-expression Genes
The approach was proposed
by Bettina Harr and Christian Schlotterer. They made use of the character of operon. An operon is a cluster of
functionally related genes regulated and transcribed as a unit [1]. Bettina
Harr et al.'s comparison method is to evaluate normalization methods
making use of the fact that bacterial genes are organized in operons. In an operon two or more
adjacent genes are co-transcribed into a single mRNA. Thus, genes located
in a given operon are expected to be highly correlated
in their expression level. This fact provides a basis for a test of which
normalization method would best predict this correlation [4].
4.5 Comparison of Different Normalization Models
As discussed in the second
part, different normalization methods deal with different problem in microarray data detection and harvest, therefore have their
advantage and disadvantage respectively. A few normalization methods can
be combined or to be performed sequentially on a dataset. However, if replicate
experiments are allowed, quantile normalization
perform relatively well on most of the data tested. Bolstad
et al. [2] compared quantile method with
some other methods, including two baseline-based methods and two methods
extended from ratio versus intensity method. Using variance comparison,
he proved that quantile performs better or at least approximately equal to
other four methods. Using bias comparison, it is shown that quantile
method produced the slope closer to 1 than any other method. Concerning
experiment cost, if replicate experiments are not available, the performance
of sole Adaptive normalization overwhelms other sole normalization methods.
In this mini-survey, we
first introduce microarray and its important role
in today's bioinformatics research. Some technical problems will induce
inaccuracy in sampling, fluorescence intensity detection, and data harvest.
Therefore, to ensure data comparison meaningful, normalization is crucial
to provide a standardization step before and functional algorithm can be
applied. We classified and introduced some common normalization methods
and evaluation approaches. Finally, for single normalization methods, according
to performance, we recommended the Quantile normalization
method if replicate experiments are available and Adaptive normalization
method if replicate experiments are not available. We also think that normalization
performance would be improved if we combine certain normalization methods
together.
[1] Rice Knowledge Bank.
www.knowledgebank.irri.org/glossary. Definition of Operon.
[2]
B. M. Bolstad, R. A. Irizarry, M. Astrand,
and T. P. Speed. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias.
Bioinformatics, 19:496{501, 2003.
[3] Z. Cai, M. Heydari,
and G. Lin. Microarray missing value imputation
by iterated local least squares. In The Fourth Asia-Pacific Bioinformatics Conference
(APBC 2006), 2005.
[4]
Bettina Harr and Christian Schlotterer. Comparison of algorithms for the analysis
of affymetrix microarray
data as evaluated by co-expression of genes in known operons.
Nucleic Acids
Research, 34, 2006.
[5] S. Oba, M. Sato,
[6] J. Quackenbush. Microarray data normalization and transformation. Nature Genetics, 32:496{501, 2002.
[7]
Gordon K. Smyth and Terry Speed. Normalization of cdna
microarray data. Methods, 2003.
[8]
Yingdong Zhao, Ming-Chung Li, and Richard Simon. An adaptive method of cdna
microarray normalization. BMC Bioinformatics, 2005.
you are the
th visitor